

Reproducing Reaction Mechanisms with Machine-Learning Models Trained on a Large-Scale Mechanistic Dataset
초록
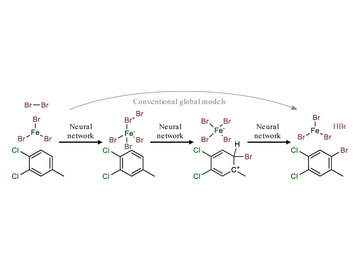
유기 반응에 대한 메커니즘적 이해는 반응 개발, 불순물 예측, 그리고 원리적으로는 반응 발견에 도움을 줄 수 있다. 여러 기계학습 모델이 반응 생성물 예측 과제를 다루어 왔지만, 이를 반응 메커니즘 예측으로 확장하는 일은 그에 상응하는 메커니즘 데이터셋의 부재로 인해 가로막혀 왔다. 이 연구에서 우리는 전문가 반응 주형(template)을 사용하여 실험적으로 보고된 반응물과 생성물 사이의 중간체를 보완함으로써 그러한 데이터셋을 구축하고, 그 결과로 얻은 5,184,184개의 기본 단계(elementary step) 데이터셋으로 여러 기계학습 모델을 학습시킨다. 우리는 이들 모델이 반응 경로를 예측하고 촉매와 시약의 역할을 재현하는 능력에 초점을 맞추어 그 성능과 역량을 살펴본다. 또한 기존 모델이 흔히 간과하는 불순물 예측에서 메커니즘 모델이 지니는 잠재력을 보인다. 끝으로 새로운 반응 유형에 대한 메커니즘 모델의 일반화 가능성을 평가하여, 데이터셋 다양성, 연속 예측, 원자 보존 위반과 관련된 과제들을 드러낸다.
Original abstract (English)
Mechanistic understanding of organic reactions can facilitate reaction development, impurity prediction, and in principle, reaction discovery. While several machine learning models have sought to address the task of predicting reaction products, their extension to predicting reaction mechanisms has been impeded by the lack of a corresponding mechanistic dataset. In this study, we construct such a dataset by imputing intermediates between experimentally reported reactants and products using expert reaction templates and train several machine learning models on the resulting dataset of 5,184,184 elementary steps. We explore the performance and capabilities of these models, focusing on their ability to predict reaction pathways and recapitulate the roles of catalysts and reagents. Additionally, we demonstrate the potential of mechanistic models in predicting impurities, often overlooked by conventional models. We conclude by evaluating the generalizability of mechanistic models to new reaction types, revealing challenges related to dataset diversity, consecutive predictions, and violations of atom conservation.